Every time a cell divides, it must copy over three billion letters of DNA with remarkable accuracy. This process is not as simple as duplicating text on a computer. The DNA is tightly packed inside the nucleus and folded into a three-dimensional structure. As the cell’s replication machinery moves along the DNA, it must navigate this crowded and highly organised landscape.
A recent study from NCBS uncovers how one of the genome’s architectural features may inadvertently become a source of DNA damage and mutations in cancer. The researchers found that sites where two key genome-organising proteins (CTCF and cohesin) bind together can create obstacles during DNA replication, which makes these regions vulnerable to damage.
CTCF and cohesin are the architects of the genome. Together, they help fold DNA into loops and domains, bringing genes and their regulatory elements into contact while keeping unrelated regions apart. These structures are essential for controlling which genes are switched on or off.
However, previous studies discovered a puzzle. The DNA regions where CTCF and cohesin bind together, known as CTCF/cohesin-binding sites (CBSs), frequently accumulate mutations in many types of cancer. Why these important architectural landmarks became mutational hotspots remained unclear.
“We know that these sites are crucial for organising the genome, but they are also among the most frequently mutated non-coding regions in cancer. We wanted to understand what makes them so vulnerable,” said Dr Faseela Elangoli Ebrahimkutty, the lead author of the study.
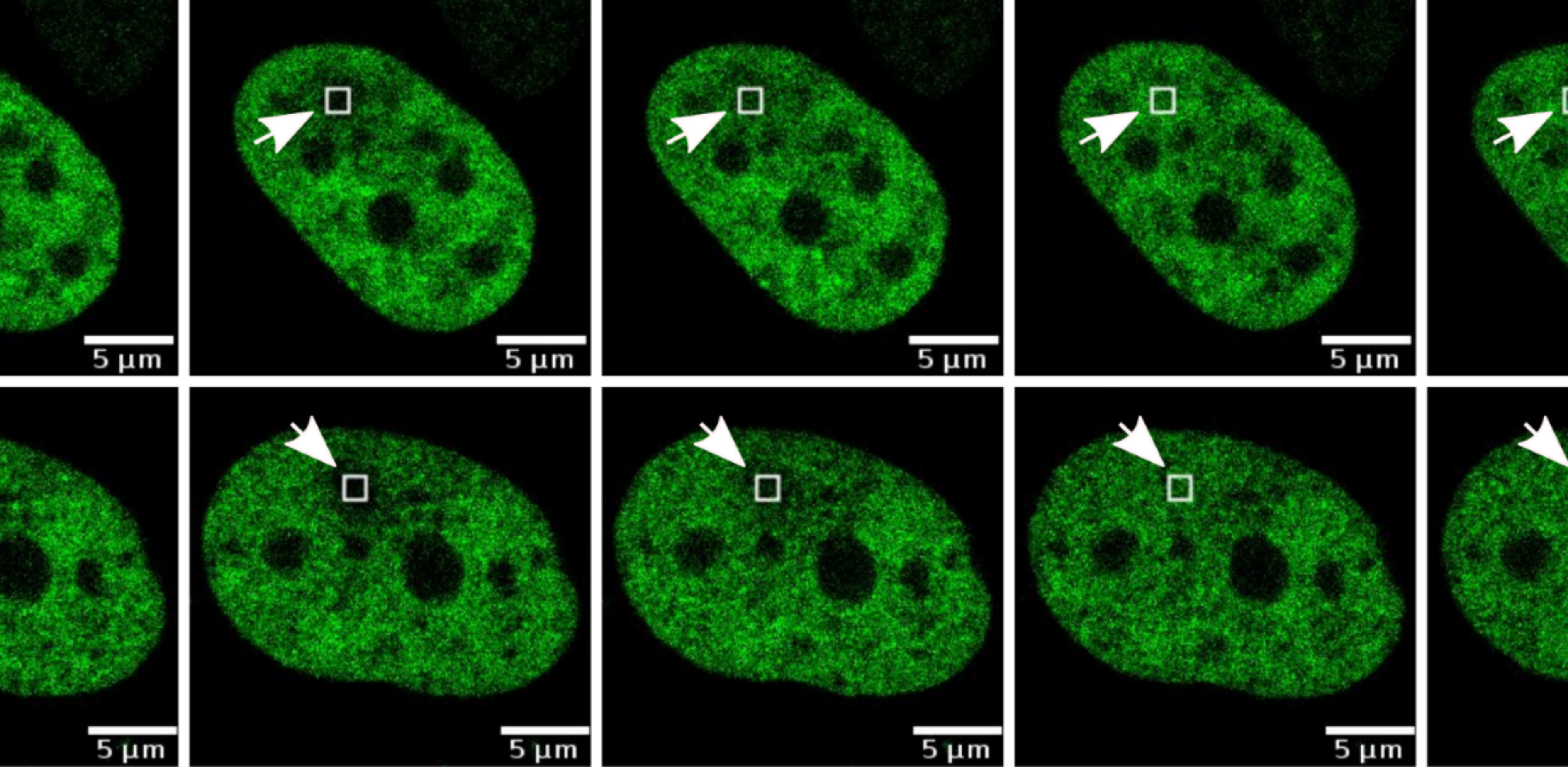
Studying DNA replication is challenging because not all cells in a population are copying their DNA at the same time. The research team temporarily halted the cell cycle and then released the cells together, creating a population in which many cells entered the DNA-copying stage at the same time. The team then focused on events that occur during replication. One possibility was that CTCF and cohesin temporarily move away when DNA is being copied, allowing replication to proceed unhindered. Instead, the team found that both proteins are likely attached to DNA throughout replication.
But, if these proteins stay attached while the replication machinery is trying to copy DNA, could they act as roadblocks?
To test this, the researchers tracked proteins that are known to appear when replication forks (the structures that copy DNA) slow down or stall. They observed that several replication stress-associated proteins, like MRE11, STN1 and FANCD2, accumulated specifically at CBSs. The stronger the binding of CTCF and cohesin at a site, the stronger the signal of replication stress.
The team then looked for signs of more severe damage. When replication stress persists, stalled replication forks can collapse, leading to DNA double-strand breaks. Using multiple markers of DNA damage and repair, the researchers found clear evidence that CBSs experience elevated levels of DNA breaks and ongoing repair.
“The enrichment of both DNA damage and repair proteins suggests that these sites are under constant pressure during replication. Cells appear to actively monitor and repair damage at these locations to maintain genome stability,” explained Dr Sabarinathan Radhakrishnan, the Principal Investigator of the study.
The researchers also asked whether this phenomenon was unique to cancer cells. By examining normal human cell lines, they found similar patterns of DNA damage response at many CBSs. This suggests that replication-associated stress at these genomic landmarks may be a normal feature of cell biology. However, in healthy cells, efficient repair mechanisms likely prevent this stress from escalating into permanent mutations.
The picture changed when the team analysed cancer genomes from patients. They focused on tumours that had lost genes involved in sensing and responding to replication-associated DNA damage, particularly MRE11 and STN1.
Cancers carrying deletions in these genes accumulated more mutations at CBSs than cancers in which the genes remained intact. The effect was especially pronounced in the stomach, oesophageal, liver and breast cancers. Importantly, nearby control regions didn’t show the same dramatic increase in mutation rates. This suggests that CBSs are uniquely susceptible when DNA damage response pathways fail.
The research team propose that the very proteins responsible for organising the genome may also create challenges for DNA replication. As replication machinery encounters CTCF and cohesin bound to DNA, it may slow down or stall. Although repair systems usually resolve these problems, defects in DNA damage response can allow errors to accumulate. Over time, these errors may contribute to the elevated mutation rates observed at CBSs in cancer.
“Understanding why certain regions of the genome are more prone to mutations helps us better interpret cancer genomes,” said Dr Radhakrishnan. “It also provides new insights into how chromatin architecture shapes the mutational landscape of cancer,” he added.
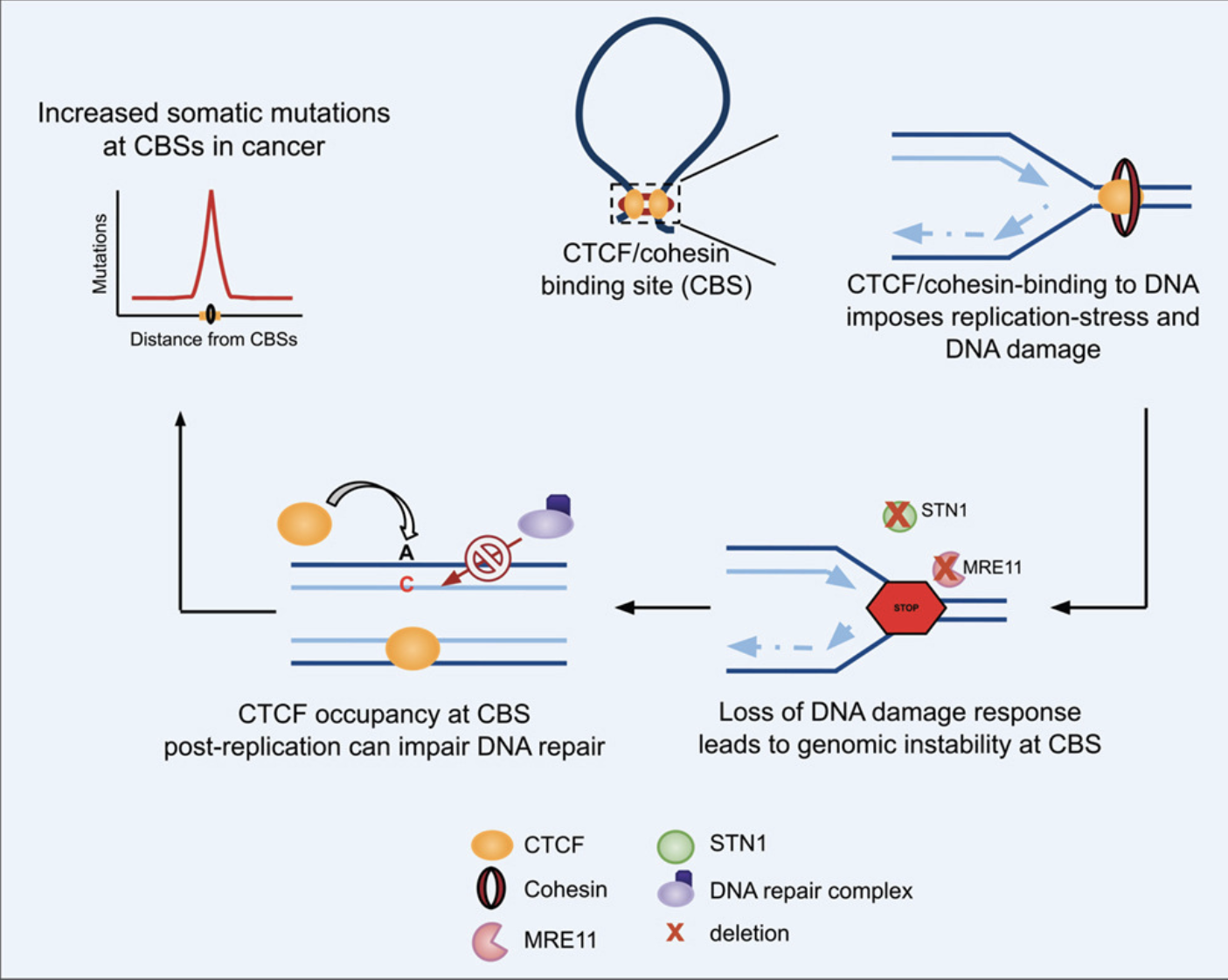
Schematic overview of the study
Link to the study- https://www.sciencedirect.com/science/article/pii/S2589004226000210


0 Comments