When clinicians first described Lowe syndrome in the 1950s, they noticed a pattern: children were born with congenital cataracts, went on to develop neurological difficulties and experienced progressive kidney problems. Decades later, scientists traced the condition to mutations in a single gene called OCRL. But how exactly a defect in this gene leads to such wide-ranging symptoms has remained an open question. Some of the symptoms have also hinted at issues with the way the cell’s powerhouses, mitochondria, function. A recent study at NCBS found that mitochondria may not function as they should in certain cell types lacking OCRL.
OCRL encodes an enzyme that modifies small lipid molecules inside cells. These lipids help organise membranes and control traffic between cellular compartments. Over the years, OCRL has been spotted in several places inside the cell, like the plasma membrane, endosomes, and other membrane-bound structures. Yet, patients with Lowe syndrome sometimes show signs and symptoms that resemble mitochondrial disorders: muscle weakness, reduced energy production, and structural abnormalities in mitochondria seen in biopsy samples. “Whether these mitochondrial changes are a direct consequence of losing OCRL, or simply a secondary effect of kidney dysfunction and altered metabolism, has been unclear,” says Dr. Ron George Philip, the lead author of the study.
Ron and team used induced pluripotent stem cells, or iPSCs, to check this. These are cells taken from somatic tissues such as blood or skin that can be reprogrammed to an early developmental state and then coaxed into becoming different cell types. Using gene editing, they created iPSCs in which OCRL was deleted. From these, they generated neural stem cells and, later, mature neural cultures. This approach allowed them to study the effects of OCRL loss in brain-related cells, independent of the complex physiological changes that occur in patients.
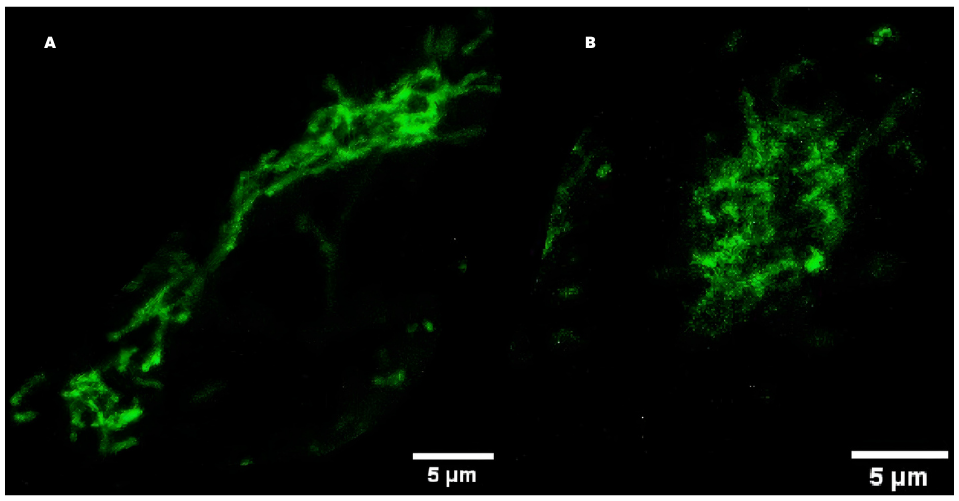
Under the microscope, the mitochondria in neural stem cells lacking OCRL looked subtly different. Instead of forming elongated, interconnected networks, they appeared slightly smaller, rounder, and less branched. These are signs of mild fragmentation. Mitochondria constantly undergo fission and fusion, balancing between breaking apart and merging together. This dynamic behaviour is closely tied to how efficiently they produce energy. Even modest shifts in this balance can reflect changes in cellular metabolism.
The research team measured mitochondrial membrane potential, a key indicator of mitochondrial health. “Mitochondria generate energy using a kind of electrical charge across their inner membrane. That charge is what powers the production of ATP, the cell’s main energy currency. In neural stem cells lacking OCRL, we consistently saw that this charge was lower,” says Ron. “What was particularly interesting is that not all the cells behaved the same way. Some were almost normal, while others showed a clear drop in membrane potential. This suggests that the response to losing OCRL isn’t uniform. In a heterogeneous neural culture, which contains a mix of different neural cell types, cells seem to cope with the loss of OCRL differently.” The team had previously found that neural stem cell cultures lacking OCRL tend to have a higher proportion of glial precursor cells. Because glial precursors rely more on non-mitochondrial sources of energy, this difference in cell type could explain why some cells were less affected than others
The research team also measured how these cells consumed oxygen and generated energy. They found modest reductions in basal respiration and ATP-linked respiration. At the same time, glycolysis (another pathway cells use to produce energy) was also slightly impaired. These changes were not catastrophic. The cells were still viable and functional. But together, they contributed to a subtle metabolic imbalance.
When the team examined the original iPSCs lacking OCRL, mitochondrial function appeared largely normal. Stem cells are known to rely more heavily on glycolysis than on mitochondrial respiration. As cells differentiate into neurons, however, they become increasingly dependent on oxidative phosphorylation. In line with this shift, OCRL-deficient neurons also showed reduced mitochondrial membrane potential, particularly as they matured. The defect, therefore, seems to arise in cells that rely more heavily on mitochondria for energy.
Could these mitochondrial changes be directly caused by altered lipid signalling? Since OCRL normally regulates levels of a lipid called PI4,5P2, the researchers tested whether reducing this lipid could rescue mitochondrial defects. Pharmacological inhibition lowered PI(4,5)P2 levels, but mitochondrial membrane potential remained unchanged. This suggests that the mitochondrial phenotype may not simply be due to elevated lipid levels, but could involve other roles of OCRL, perhaps through protein - protein interactions or indirect effects on organelle communication.
A broad analysis of gene expression revealed only modest changes in mitochondria-related genes. Rather than a dramatic rewiring of the mitochondrial transcriptome, the data suggest subtle, coordinated shifts. These are small changes that, collectively, may influence how mitochondria behave in OCRL-deficient cells.
“This study highlights the power of modern stem cell technology in understanding the cell biological basis of human diseases. It is possible to propose specific hypotheses and test them in a laboratory setting, typically not possible with human patients. A clear understanding of disease mechanism is the foundation on which potential treatments can be formulated, “ says Prof. Raghu Padinjat, the principal investigator of the study.


0 Comments