Using maths to correct for natural selection in Mutation Accumulation experiments
Lindi was delivering a talk on a study that showed how a good number of mutations from an MA experiment (mutation accumulation) were beneficial to the bacteria carrying them, when she received a perplexing question from the audience: “What if you are seeing more beneficial mutations because of selection during the MA experiment?”. Let’s understand Lindi’s perplexity with a little background.
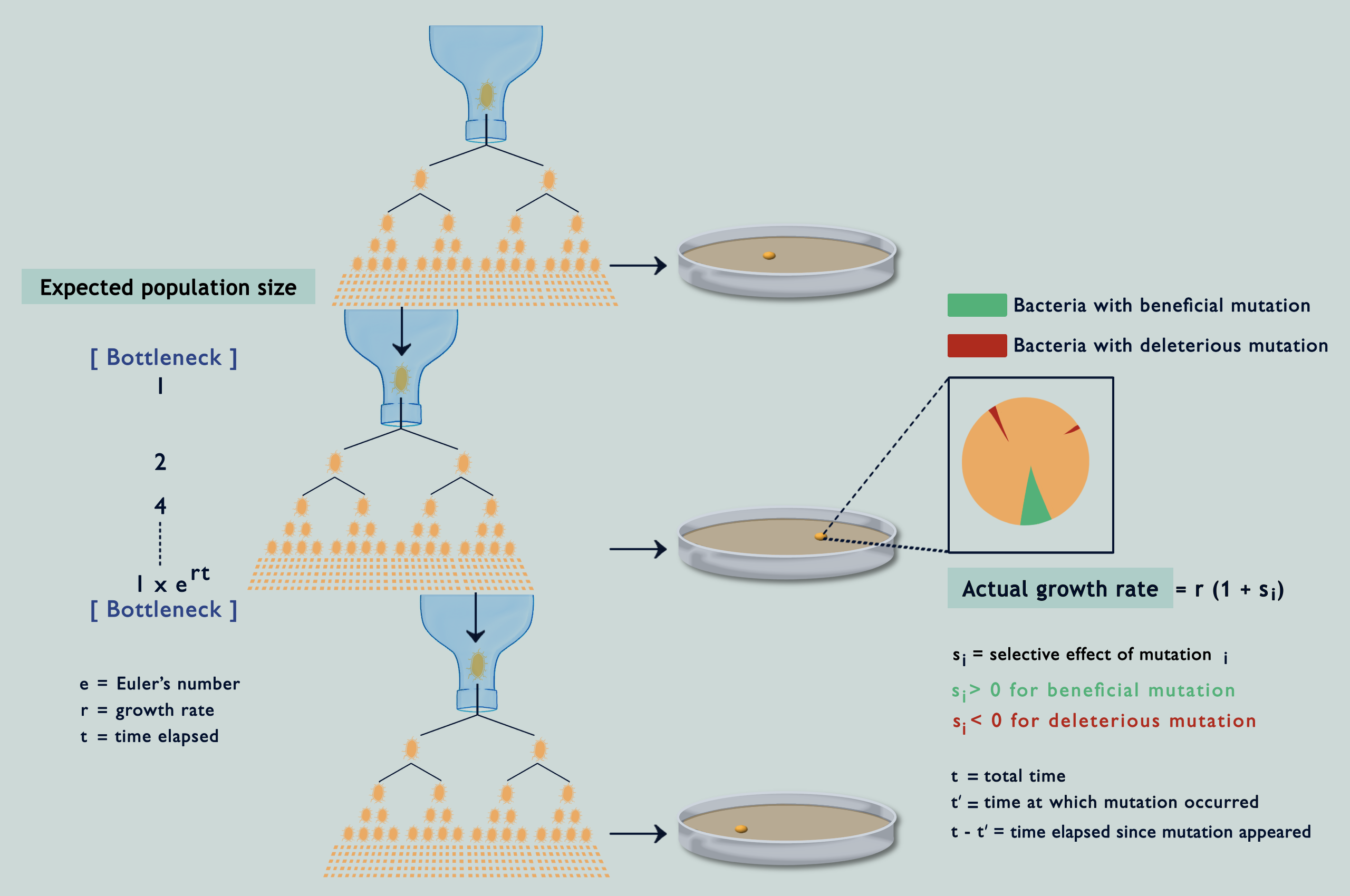
Suppose you wanted to characterise mutations occurring in a population. You might go about randomly sampling some individuals and investigating their genomes, but only to end up with a skewed view of the desired results. This is due to natural selection: some (beneficial) mutations make individuals ‘fitter’ or capable of producing more offspring than others, while other (deleterious) mutations have the opposite effect. Consequently, you see more beneficial mutations than deleterious ones in your sample, even if both occur at the same rate. To get an unbiased representation of occurring mutations, you must somehow reduce the effect of natural selection. MA experiments have been the gold standard for this since the 1920s. They enable estimation of the rates of different mutations and how they impact the fitness of individuals carrying them (fitness being a quantitative estimate of reproductive success). The protocol involves isolating individuals at each generation and letting them thrive in an environment with abundant resources, or in other terms, by passing the population through a series of single-individual ‘bottlenecks’ (refer image above). Because of this, individuals making through the bottlenecks have no one to compete with and get to propagate their lineage unrestrainedly with all of their unique mutations (the easiest way to top your class is to have no classmates!).
On being asked about the possibility of selection during MA, Lindi and her collaborator, Deepa, did some simulations, and lo and behold! As the bacteria divided and accumulated mutations in silico, selection was caught tampering with their fates! But, how come? What I didn’t mention earlier is that while separating individuals at each generation is usually possible for larger organisms, the same isn’t true for microbes. A bacterium for example must be allowed to reproduce for at least 15 generations on an agar plate before it can grow into a colony big enough to be seen. Only then can we transfer one bacterium from it to a lavishly big agar plate. It has always been assumed that these windows of 15 generations, being like the blink of an eye in evolutionary time, aren’t enough for selection to tinker with the accumulating mutations; but the simulations were telling a different story (refer image above).
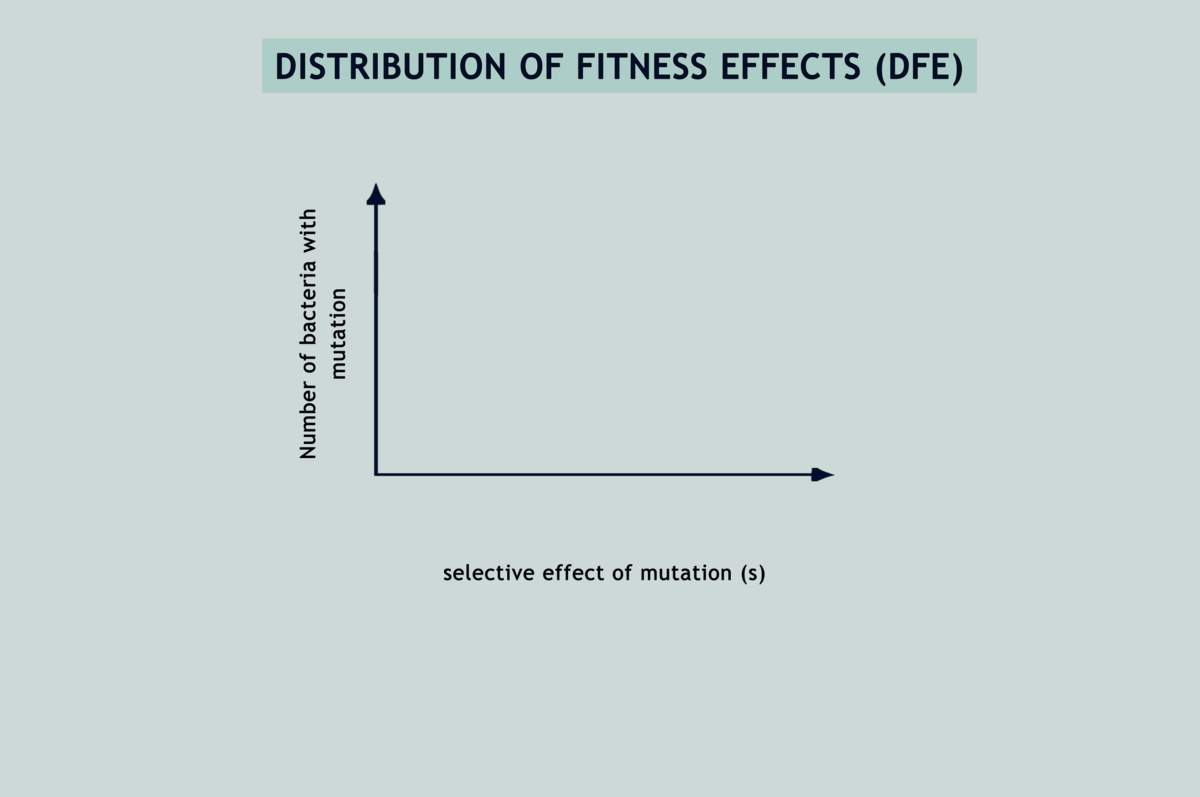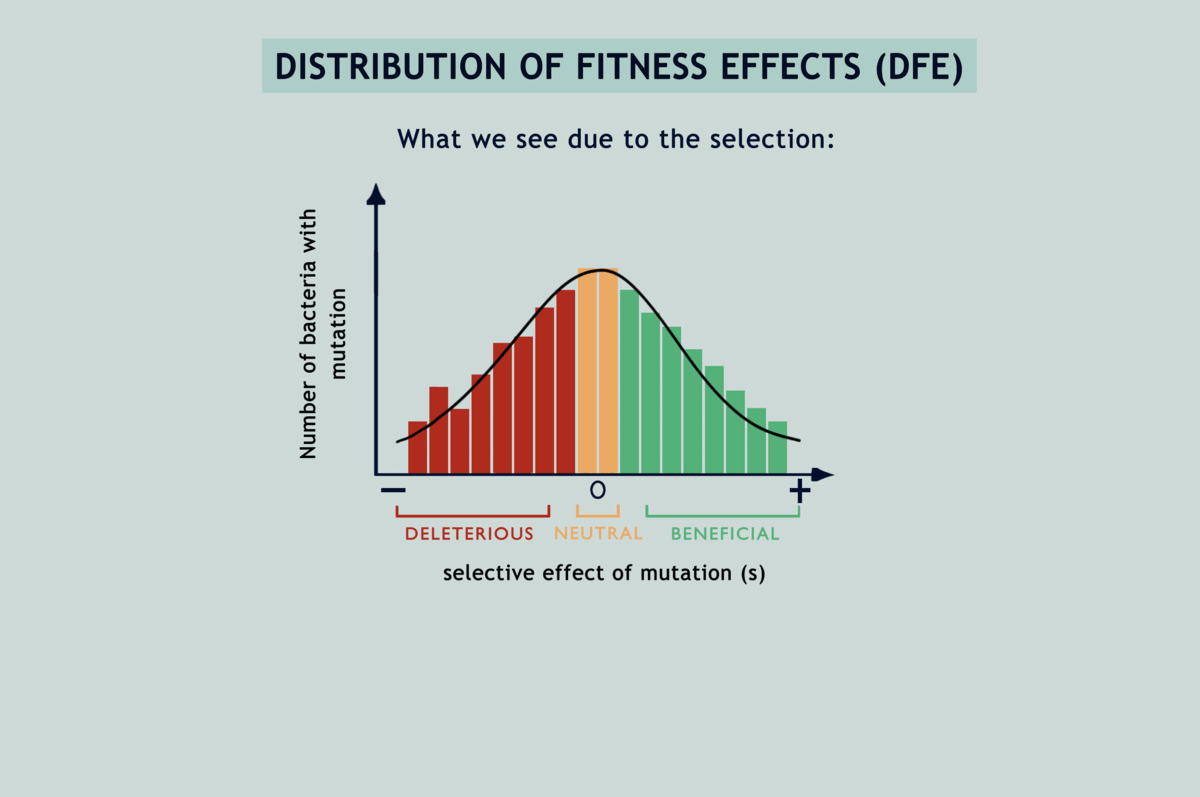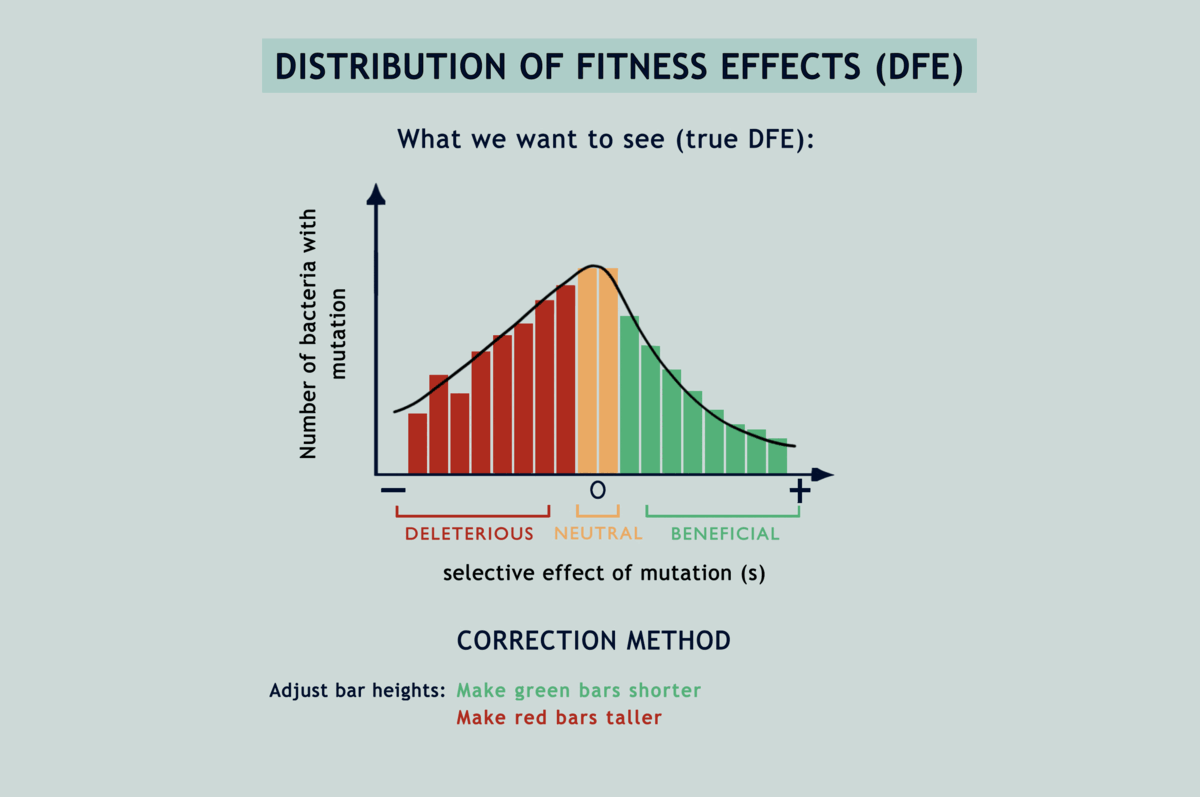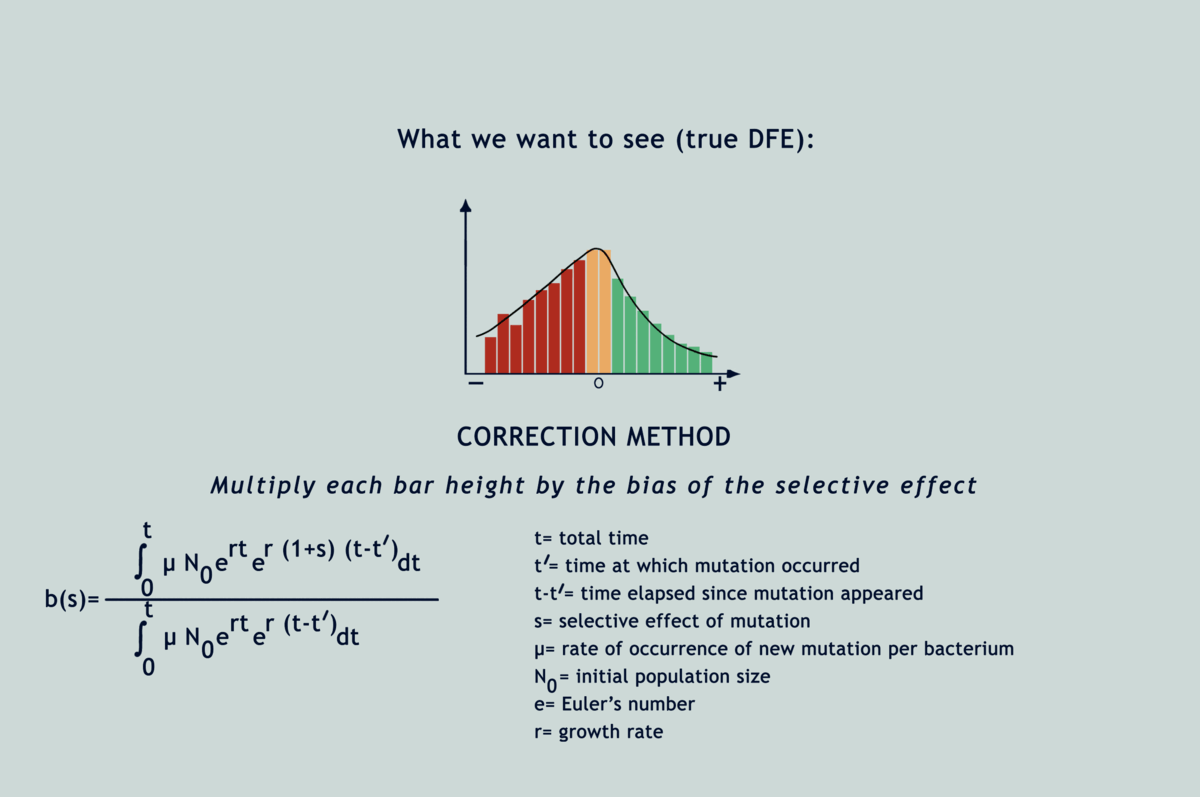
Understanding the statistical distribution of the effects of mutations (represented as a histogram known as a DFE or distribution of fitness effects) forms the basis of many models of evolution and adaptation. The bias imposed by selection during microbial MA experiments alters the shape of underlying DFEs, throwing interpretations derived from them way off the chart (refer GIF below). Selection has been acting in between the bottlenecks and slyly fooling researchers, as if in a game of peek-a-boo, until they finally exclaimed, ‘I see you!’; but now what? Previous attempts to correct for the selection bias have been scanty and applicable only to deleterious mutations. Lindi and Deepa worked out a correction method that applies to both deleterious and beneficial mutations. They also figured the same for when mutations occur in one environment but their impacts on fitness are examined in another. By applying this correction to E.coli growing on LB agar, their estimate of the fraction of mutations that are beneficial came down from 40% to as low as 5.8%. Occurrence rates fell for beneficial mutations and rose for deleterious ones, while observed DFEs started mimicking expected ones. The underlying mathematics is surprisingly simple for what it achieves (refer GIF below).
For details, refer to the study here.
So, has this bias been plaguing all microbial MA experiments till date? Likely. However, before Deepa’s team took up the challenge (Sane et al., 2018), people rarely made efforts to characterise single mutations obtained through MA. Fitness is generally averaged over multiple mutations, so an overwhelming number of beneficial mutations, even if they occur, remain undercover. For such studies, the selection bias might not cause a noticeable shift in the DFE. The bias correction method becomes important in case of DFEs for single mutations. It can be useful to also think of a way to correct for selection in the background of multiple mutations. The maths for that is yet to be worked out. At least till then, if asked again about selection during MA experiments, Lindi and Deepa have an answer right up their sleeves!
Acknowledgements:
I’m grateful to Dr. Lindi M. Wahl and Dr. Deepa Agashe for letting me write about their work and suggesting improvements to the article. I also thank Upasana Sardar for materialising my vision for the image and GIF (check out her digital art gallery here).


0 Comments