
Proteins drive nearly every process within a cell. They are made of long chains of amino acids that fold into precise three-dimensional shapes, enabling each protein to carry out its specific function. By studying a protein’s 3D structure, scientists can deduce its role in the cell.
While advances in sequencing have given us hundreds of millions of protein sequences, only a small fraction have known structures. That’s because determining 3D structures is still a slower, more resource-intensive task, relying on complex computational models and sophisticated experimental techniques.
However, without 3D structural data, how can we uncover the functions of the vast majority of proteins?
A recent study led by Prof R. Sowdhamini at NCBS developed a database to help predict protein function, without the availability of information on its 3D structures. The Genomic Distribution of Superfamilies, or the GenDiS3 database, achieves this by analyzing conserved protein sequences. Conserved protein sequences are parts of a protein that are very similar across different organisms and perform similar important functions.
Researchers first used bioinformatic tools to search through a massive database of all protein sequences described to date, to look for subtle similarities between the reference proteins and unknown sequences.
GenDiS3 compares unknown protein sequences to a library of well-characterised protein domains-which are distinct structural and functional units within proteins- from the SCOPe database (a database that classifies protein structures).
If an unknown sequence shares these conserved elements with a known protein domain, the function is likely to be similar.They then verified these matches using Hidden Markov Models(HMM’s)- statistical tools which can detect even faint evolutionary links between proteins.
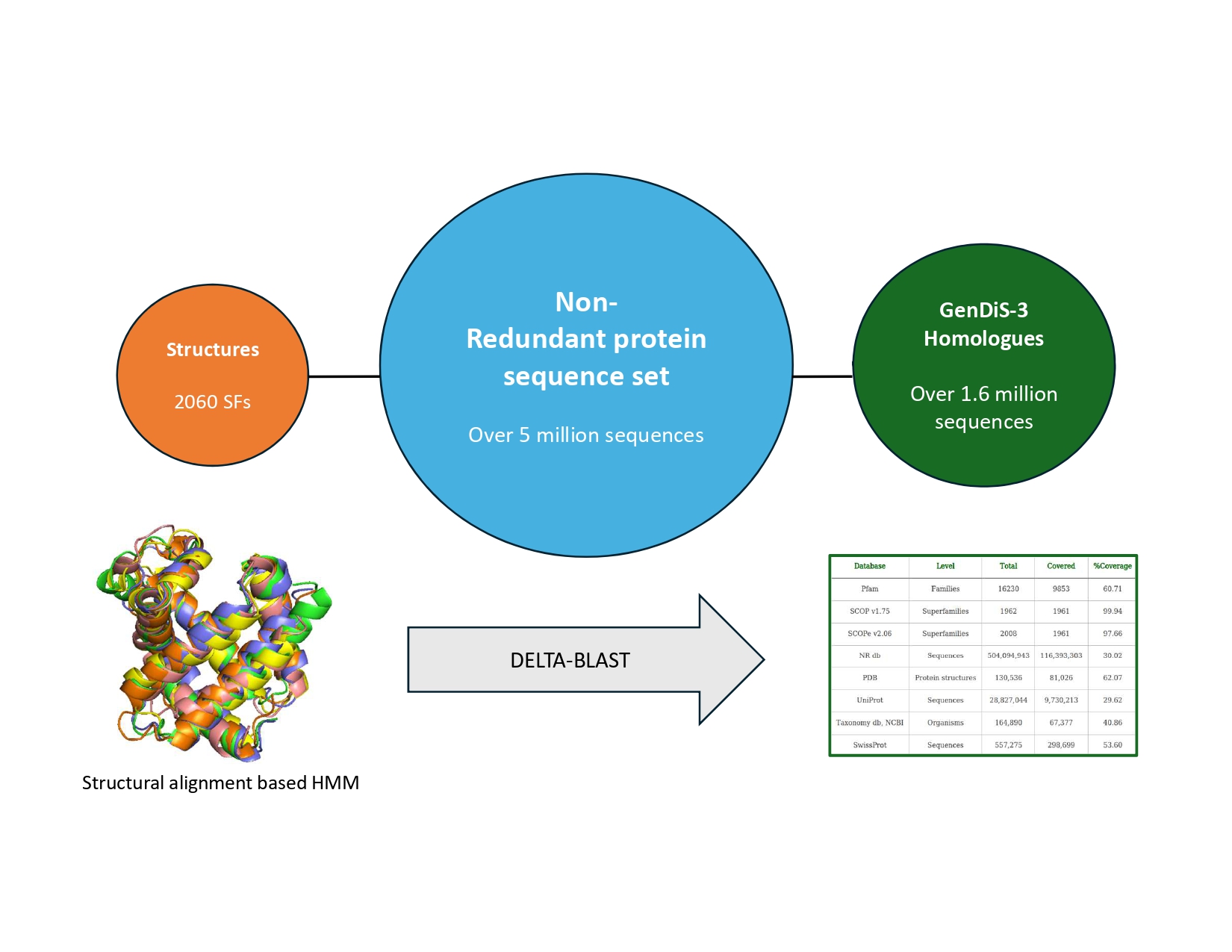
Using this advanced computational pipeline, the team identified and validated over 116 million trusted homologous sequences—those that are evolutionarily connected and potentially functionally similar, corresponding to roughly 23% of the entire public sequence database.
“By providing a robust framework to link sequences to known structures, our database empowers researchers to make more accurate predictions about a protein's function, even in the complete absence of an experimental structure”, said Sarthak Joshi, lead author of the study.
By systematically mapping millions of protein sequences to known domain superfamilies using powerful computational tools, GenDiS3 bridges the gap between the flood of genetic data and limited structural information.
Through a case study on the glycolysis pathway (the process by which cells break down glucose for energy), the researchers used the GenDiS3 database to trace how proteins of this pathway have evolved in different organisms. In another case study, they analysed plant-related proteins and identified important functions that had not been described earlier, showing that this database can be used to analyse and predict functions of genes of agricultural importance.
The database thus reveals how common protein building blocks are reused and modified in different organisms, helping trace the evolutionary processes that have enabled proteins to adapt to diverse biological roles.
“The world of sequence data is growing at an exponential rate, and a primary challenge will be to ensure our database can scale accordingly. Our future efforts will focus on optimising our computational pipeline to handle this data influx more efficiently, potentially by reducing the need for certain validation steps and improving data management. The continued development and expansion of the GenDiS3 database will be essential to keep pace with the rapid advancements in genomics, ensuring it remains a critical and relevant tool for researchers working to understand the fundamentals of protein function and evolution,” said Joshi.
“There are far more proteins with sequence information than those of structures, but do in fact adopt known folds. Starting from the protein structural end, GenDis3.0 database is the outcome of a one-time computational exercise to bridge this gap, accept significant connections and to permit the users to extract this information conveniently. The users can follow either structural hierarchy (fold, superfamily and family) or taxonomic hierarchy (like kingdom, phylum, class, order, family) to extract protein homologues from the sequence space. Early realisation of homologues of a protein family can enable design of experiments for biologists.” said Prof R. Sowdhamini, corresponding author of the study.


0 Comments